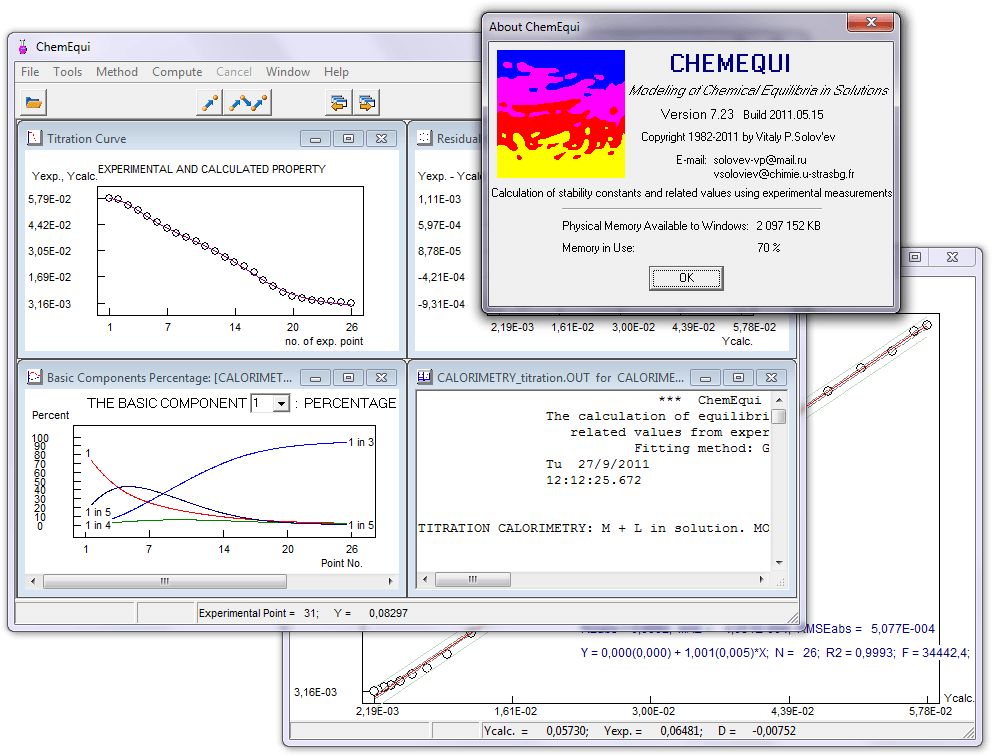
Программа ChemEqui рассчитывает константы химических равновесий и сопутствующие параметры, исходя из экспериментальных результатов физико-химических методов, таких как УФ, ИК и ЯМР спектроскопия, калориметрия, потенциометрия и кондуктометрия. Константы равновесий рассчитываются методом наилучшей подгонки экспериментальных данных к расчетным величинам для выбранной (предполагаемой) модели равновесий (равновесных реакций).
The ChemEqui program realizes the method of non-linear least-squares for computations of equilibrium constants and related quantities from experimental results of UV/Vis, IR and NMR spectroscopy, calorimentry, potentiometry and conductometry. The equilibrium constants are calculated by best-fitting of the experimental data with an estimated chemical model of the equilibrium system.
В ChemEqui используются три подхода для поиска минимума системы нелинейных уравнений методом наименьших квадратов: метод Ньютона-Гаусса с аналитическим представлением производных (высокая скорость расчетов), симплекс алгоритм и метод Монте-Карло. Это позволяет существенно улучшить надежность расчетов равновесных констант.
Использование ChemEqui
Распаковать файл архив содержащий директорию ChemEqui. Рекомендуется использовать несистемный диск для директории ChemEqui в случае Windows 7 и Windows Vista.
CHemEqui uses three algorithms of minimization to solve non-linear least squares problems: Gauss—Newton with analytical presentation of derivatives, Simplex and Monte Carlo allowing one to significantly improve the reliability of the stability constant calculations.
Using ChemEqui
Unpack the archive containing the directory the ChemEqui program. For Windows 7 and Windows Vista, the utilization of non-system disk for the ChemEqui directory is preferable.
Литература
References
Solov’ev, V.P.; Vnuk, E.A.; Strakhova, N.N.; Raevsky, O.A.; Thermodynamics of the Complexation of Salts of Alkali and Alkali-Earth Metals with Cyclic Polyethers. Itogi Nauki I Techniki. Seriya Khimicheskaya Termodinamika i Ravnovesiya (Rus. book), 1991, 7, VINITI, Moscow, 374 pp.
Solov’ev, V.P.; Baulin, V.E.; Strakhova, N.N.; Kazachenko, V.P.; Belsky, V.K.; Varnek, A.A.; Volkova, T.A.; Wipff, G.; Complexation of Phosphoryl- Containing Mono-, Bi- and Tri-Podands with Alkali Cations in Acetonitrile. Structure of the Complexes and Binding Selectivity. J. Chem. Soc. Perkin Trans. 2, 1998, 1489-1498.
Georg S., Billard I., Ouadi A., Gaillard C., Petitjean L., Picquet M., Solov’ev V. Determination of Successive Complexation Constants in an Ionic Liquid: Complexation of UO22+ with NO3- in C4-mimTf2N Studied by UV-Vis Spectroscopy. J. Phys. Chem. B, 2010, 114, No. 12, 4276–4282.
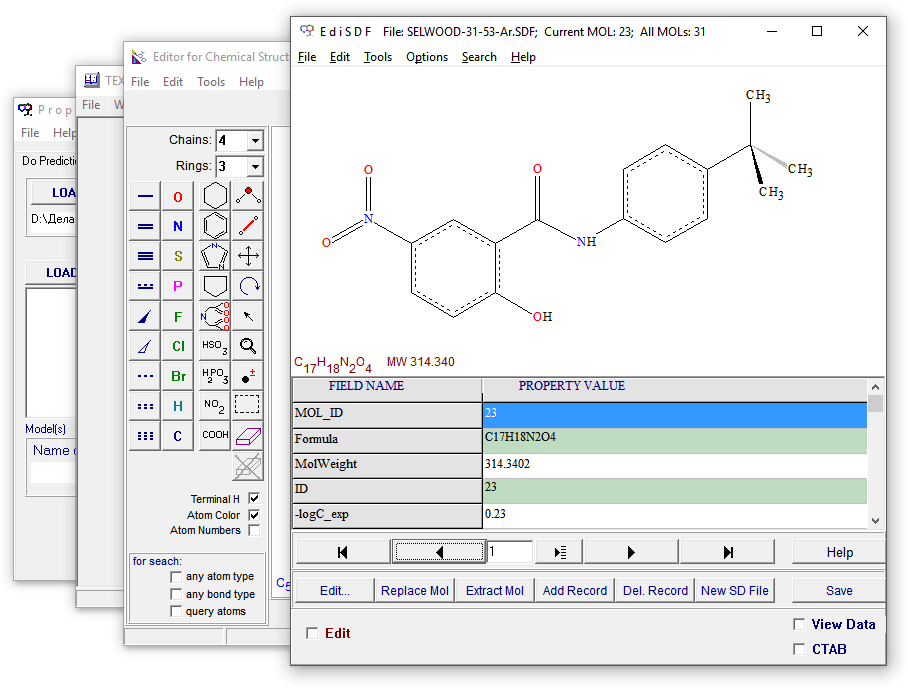
Редактор EdiSDF предназначен для визуализации, управления и редактирования файлов, содержащих химические 2D и 3D формулы в формате Structure-Data Files (SDF).
The editor EdiSDF provides visualization, management and edition of Structure-Data Files (SDF) of chemical 2D and 3D formulae.
Редактор EdiSDF является частью проекта ISIDA. Проект ISIDA — совместный проект между проф. А. Варнеком (Лабораторией химической информатики, UMR 7177 CNRS, Universite de Strasbourg, 4, rue B.Pascal, Strasbourg, 67000, France) и д.х.н. в.н.с. Соловьевым В.П. (Лаборатория новых физико-химических проблем, зав. академик Цивадзе А.Ю., Институт физической химии и электрохимии, РАН, 119991 Москва, Ленинский пр., 31а).
Редактор EdiSDF включает:
Редактор EdChemS химических 2D формул. Windows 32-bit;
Программу CombiLib для генерации виртуальных химических соединений (библиотек);
Программу FMF для предсказания физических, химических и биологических свойств с использованием готовых моделей ISIDA/QSPR;
Распаковать архив содержащий директорию редактора EdiSDF. Программа не требует инсталяции.
The EdiSDF program is a part of the ISIDA project. ISIDA is a collaborative project between the Laboratory of Chemoinformatics by Prof. Alexandre Varnek (Laboratoire d’Infochimie, UMR 7177 CNRS, Universite de Strasbourg, 4, rue B.Pascal, Strasbourg, 67000, France) and Dr. Vitaly Solov’ev (Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninskiy prospect, 31a, 119991, Moscow, Russian Federation).
EdiSDF includes:
The EdChemS editor of chemical 2D formulae. Windows 32-bit;
The CombiLib program for a generation of virtual chemical compounds (Libraries);
The FMF program for predictions of physical, chemical and biological properties using developed ISIDA/QSPR models;
Unpack the archive containing the directory the EdiSDF program.
Литература
References
Solov’ev, V.P., Kireeva, N.V., Tsivadze, A.Y., Varnek, A.A. Structure-Property Modelling of Complex Formation of Strontium with Organic Ligands in Water. J. Struct. Chem. 47(2), 298-311 (2006).
Varnek, A., Fourches, D., Hoonakker, F., Solov’ev, V.P. Substructural Fragments: An Universal Language to Encode Reactions, Molecular and Supramolecular Structures. J. Comp.-Aided Mol. Design 19(9-10), 693-703 (2005).
Varnek, A., Fourches, D., Horvath, D., Klimchuk, O., Gaudin, С., Vayer, P., Solov’ev, V., Hoonakker, F., Tetko, I.V., Marcou, G. ISIDA – Platform for Virtual Screening Based on Fragment and Pharmacophoric Descriptors. Cur. Computer-Aided Drug Design 4(3), 191-198 (2008).
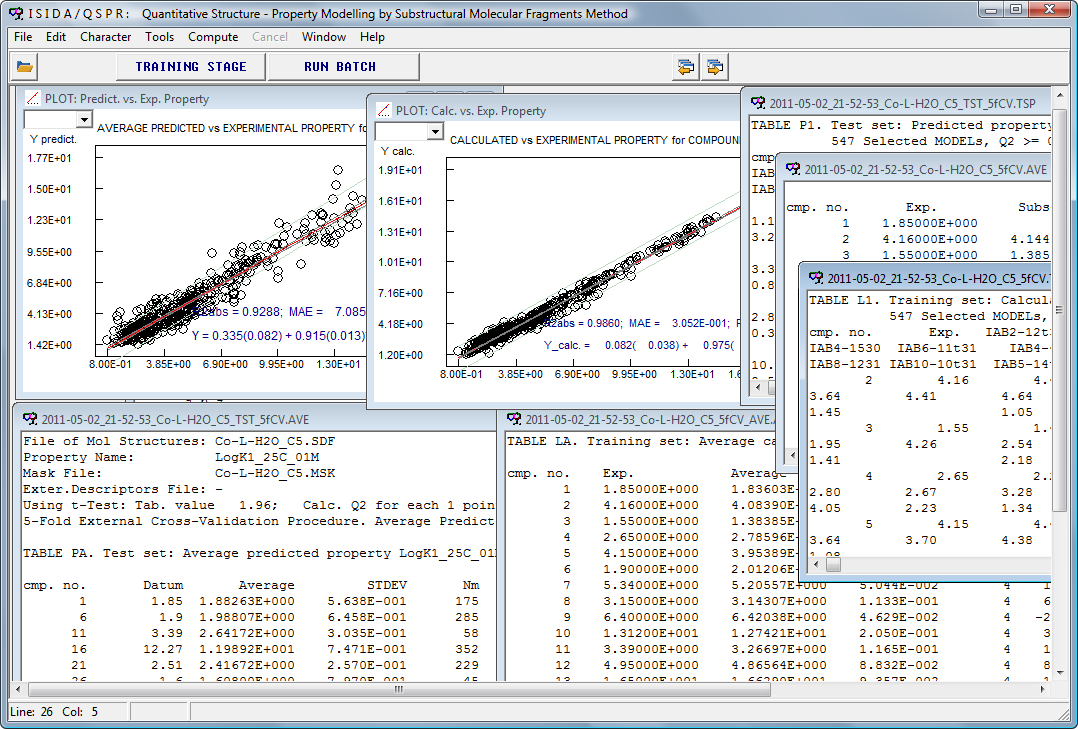
В программе ISIDA/QSPR используются субструктурные молекулярные фрагменты (SMF) и многомерный линейный регрессионный анализ (MLR) для QSPR и QSAR моделирования и предсказания физических, химических и биологических свойств.
The ISIDA/QSPR program realizes Multiple Linear Regression Analysis (MLR) and Substructural Molecular Fragments (SMF) for QSPR and QSAR modelling and prediction of physical, chemical and biological properties.
В качестве исходных данных в ISIDA/QSPR служат известные экспериментальные величины моделируемого свойства для ряда соединений обучающего набора данных. Субструктурные молекулярные фрагменты как подграфы молекулярных графов соединений применяются в качестве дескрипторов (независимых переменных) в QSPR моделях. Как правило, применяются кратчайшие топологические пути. Кратность фрагмента в соединении — величина дескриптора. Дескрипторы вычисляются исключительно из данных о структурной (2D) формуле соединения.
Привлекается оригинальная комбинированная пошаговая техника увеличения и уменьшения числа SMF дескрипторов в модели для выбора переменных из их исходного множества, обеспечивающих наилучшее предсказание свойства.
ISIDA/QSPR генерирует множество устойчивых MLR моделей, каждая их которых соответствует применяемому типу SMF дескрипторов и методам ступенчатого отбора переменных. Для надежного предсказания свойств используется консенсус модель. Консенсус модель объединяет предсказания множества индивидуальных моделей. Программа рассчитывает свойство как арифметическое среднее величин, вычисленных с помощью наиболее устойчивых индивидуальных моделей, исключая выпадающие величины и применяя методы по оценке области применимости каждой индивидуальной модели.
Программа ISIDA/QSPR является частью проекта ISIDA. Проект ISIDA — совместный проект между проф. А. Варнеком (Лабораторией химической информатики, UMR 7177 CNRS, Universite de Strasbourg, 4, rue B.Pascal, Strasbourg, 67000, France) и д.х.н. в.н.с. Соловьевым В.П. (Лаборатория новых физико-химических проблем, зав. академик Цивадзе А.Ю., Институт физической химии и электрохимии, РАН, 119991 Москва, Ленинский пр., 31а).
Программа ISIDA/QSPR включает:
Редактор EdiSDF, предназначенный для визуализации и редактирования файлов, содержащих химические 2D и 3D формулы в формате Structure-Data Files (SDF). Входные данные для программы ISIDA/QSPR представлены файлами SDF.
Программу FMF для предсказания физических, химических и биологических свойств с использованием готовых моделей ISIDA/QSPR.
Модуль MolFrag для анализа субструктурных молекулярных фрагментов (SMF) и их вкладов.
Распаковать архив содержащий директорию ISIDA_QSPR. Для Windows 7 и Windows Vista строго рекомендуется размещать директорию ISIDA_QSPR не на системном диске. Программа не требует инсталляции. Файл помощи ISIDA_QSPR_Manual.doc находится в директории ISIDA_QSPR.
As initial data, ISIDA/QSPR uses known experimental values of modelling property for training set of chemical compounds. Substructural molecular fragments as subgraphs of molecular graphs of the compounds are descriptors (independent variables) in QSPR models. As a rule, shortest topological paths are applied. A fragment occurrence is a descriptor value. The descriptors are derived solely from 2D chemical structures.
Original combined forward and backward stepwise techniques are applied for selections of the most pertinent variables from initial pools of the SMF descriptors.
ISIDA/QSPR generates many MLR models; each of them corresponds to applied type of the SMF descriptors and the stepwise techniques. For reliable predictions of the properties, a consensus model is used. The consensus model combines the predictions issued from many individual models. The program computes the property as an arithmetic mean of values obtained with a collection of selected on training stage individual models excluding those leading to outlying values, and taking into account an applicability domain of each individual model.
The ISIDA/QSPR program is a part of the ISIDA project. ISIDA is a collaborative project between the Laboratory of Chemoinformatics by Prof. Alexandre Varnek (Laboratoire d’Infochimie, UMR 7177 CNRS, Universite de Strasbourg, 4, rue B.Pascal, Strasbourg, 67000, France) and Dr. Vitaly Solov’ev (Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninskiy prospect, 31a, 119991, Moscow, Russian Federation).
ISIDA/QSPR includes:
The EdiSDF editor for visualization and edition of Structure-Data Files (SDF) of chemical 2D and 3D formulae. SDF is data input format for the ISIDA/QSPR program.
The FMF program for predictions of physical, chemical and biological properties using developed ISIDA/QSPR models.
The MolFrag tools for the analysis of substructural molecular fragments (SMF) and their contributions.
Unpack the archive containing the directory of the ISIDA_QSPR program. For Windows 7 and Windows Vista, it is strongly recommended to use of non-system disk for the ISIDA_QSPR directory. See ISIDA_QSPR_Manual.doc as help file inside the ISIDA_QSPR directory.
Solov’ev V., Oprisiu I., Marcou G., Varnek A. Quantitative Structure_Property Relationship (QSPR) Modeling of Normal Boiling Point Temperature and Composition of Binary Azeotropes. Ind. Eng. Chem. Res., 2011, 50, No. 24, pp 14162–14167.
Varnek A., Solov’ev V. Quantitative Structure-Property Relationships in solvent extraction and complexation of metals. Rev. in Book: Ion Exchange and Solvent Extraction, A Series of Advances. Vol. 19, P. 319-358. A. K. Sengupta and B. A. Moyer, Eds., CRC Press, Taylor and Francis Group: Boca Raton, 2009, 679 pp.
Solov’ev, V. P.; Varnek, A. A.; Wipff, G. Modelling of Ion Complexation and Extraction Using Substructural Molecular Fragments. J. Chem. Inf. Comput. Sci., 2000, 40, P. 847-858.
Varnek, A. A.; Wipff, G.; Solov’ev, V. P. Towards an Information System on Solvent Extraction. J. Solvent Extr. Ion. Exch., 2001, 19, No. 5, P.791-837.
Varnek, A. A.; Wipff, G.; Solov’ev, V. P., Solotnov A.F. Assessment of The Macrocyclic Effect for The Complexation of Crown-Ethers with Alkali Cations Using the Substructural Molecular Fragments Method. J. Chem. Inf. Comput. Sci., 2002, 42, No. 4, P. 812-829.
Solov’ev, V. P.; Varnek, A. Anti-HIV Activity of HEPT, TIBO and Cyclic Urea Derivatives: Structure-Property Studies, Focused Combinatorial Library Generation and Hits Selection Using Substructural Molecular Fragments Method. J. Chem. Inf. Comp. Sci., 2003, 43, No. 5, P. 1703-1719.
Katritzky, A.R.; Fara, D.C.; Yang, H.; Karelson, M.; Suzuki, T.; Solov’ev, V.P.; Varnek A. Quantitative Structure-Property Relationship Modeling of ?-Cyclodextrin Complexation Free Energies. J. Chem. Inf. Comput. Sci. 2004, 44, No. 2, 529-541.
Varnek, A.; Fourches, D.; Solov’ev, V. P.; Baulin, V. E.; Turanov, A. N.; Karandashev, V. K.; Fara, D.; Katritzky, A. R. «In Silico» Design of New Uranyl Extractants Based on Phosphoryl-Containing Podands: QSPR Studies, Generation and Screening of Virtual Combinatorial Library and Experimental Tests. J. Chem. Inf. Comput. Sci., 2004, 44, No. 4, 1365-1382.
Solov’ev, V. P.; Varnek, A. A. Structure-Property Modeling of Metal Binders Using Molecular Fragments. Russ. Chem. Bull., Internat. Edit. (in Russ.: Izv. Akad. Nauk. Ser. Khim., 2004, No. 7, pp. 1380-1391) 2004, 53, 1434-1445.
Varnek, A.; Solov’ev, V. P. «In Silico» Design of Potential Anti-HIV Actives Using Fragment Descriptors. Combinatorial Chem. High Throughput Screening, 2005, 8, No. 5, 403-416.
Varnek, A.; Fourches, D.; Hoonakker, F.; Solov’ev, V. P. Substructural fragments: an universal language to encode reactions, molecular and supramolecular structures. J. Computer-Aided Mol. Design, 2005, 19, 693-703.
Katritzky, A. R.; Kuanar, M.; Fara, D. C.; Karelson, M.; Acree, W. E. Jr.; Solov’ev, V. P.; Varnek, A. QSAR modeling of blood:air and tissue:air partition coefficients using theoretical descriptors. Bioorg. Med. Chem.,2005, 13, 6450-6463.
Tetko, I. V.; Solov’ev, V. P.; Antonov, A. V.; Yao, X.; Doucet, J. P. Fan, B.; Hoonakker, F.; Fourches, D.; Jost, P.; Lachiche, N.; Varnek, A. Benchmarking of Linear and Nonlinear Approaches for Quantitative Structure-Property Relationship Studies of Metal Complexation with Ionophores. J. Chem. Inf. Model., 2006, 46, No. 2, 808-819.
Katritzky, A. R.; Dobchev, D. A.; Fara, D. C.; Hur, E.; Tamm, K.; Kurunczi, L.; Karelson, M.; Varnek, A.; Solov’ev, V. P. Skin Permeation Rate as a Function of Chemical Structure. J. Med. Chem., 2006, 49, No. 11, 3305-3314.
Katritzky, A. R.; Kuanar, M.; Slavov, S.; Dobchev, D. A.; Fara, D. C.; Karelson, M.; William, E.; Acree, W. E. Jr.; Solov’ev, V. P.; Varnek, A. Correlation of Blood — Brain Penetration Using Structural Descriptors. Bioorg. Med. Chem., 2006, 14, No. 14, 4888-4917.
Solov’ev, V. P.; Kireeva, N. V.; Tsivadze, A. Yu.; Varnek, A. A. Structure-Property Modeling of the Complexation of Strontium with Organic Ligands in Water. Zh. Structur. Khimii (Rus.), 2006, 47, No. 2, 303-317.
Varnek, A.; Fourches, D.; Sieffert, N.; Solov’ev, V. P.; Hill, C.; Lecomte, M. QSPR Modeling of the AmIII / EuIII Separation Factor: How Far Can We Predict? Solv. Extr. Ion Exch., 2007, 25, No. 1, P. 1-26.
Varnek, A.; Kireeva, N.; Tetko, I. V.; Baskin, I. I.; Solov’ev, V. P. Exhaustive QSPR Studies of Large Diverse Set of Ionic Liquids: How Accurately Can We Predict the Melting Point? J. Chem. Inf. Model., 2007, 47, No. 3, P. 1111-1122.
Horvath D., Bonachera F., Solov’ev V., Gaudin C., Varnek A. Stochastic versus Stepwise Strategies for Quantitative Structure — Activity Relationship Generations. — How Much Effort May the Mining for Successful QSAR Models Take? J. Chem. Inf. Model., 2007, 47, No. 3, P. 927-939.
Varnek A.; Fourches D.; Solov’ev V.; Klimchuk O.; Ouadi A.; Billard I. Successful «In Silico» Design of New Efficient Uranyl Binders. Solv. Extr. Ion Exch., 2007, 25, No. 4, P. 433-462.
Varnek A., Fourches D., Horvath D., Klimchuk O., Gaudin С., Vayer P., Solov’ev V., Hoonakker F., Tetko I. V., Marcou G. ISIDA — Platform for Virtual Screening Based on Fragment and Pharmacophoric Descriptors. Cur. Computer-Aided Drug Design, 2008, 4, No. 3, P. 191-198.
Varnek A., Fourches D., Kireeva N., Klimchuk O., Marcou G., Tsivadze A., Solov’ev V. Computer-Aided Design of New Metal Binders. Radiochim. Acta, 2008, 96, P. 505-511.